Blog: Quantitative Genetics and GWAS; How Genome-Wide Association Studies Are Mapping the Genetic Architecture of Every Trait That Matters in Cannabis.
- May 10
- 6 min read
Published 10AM EST, Mon May 11, 2026.
The Gap Between What We Grow and What We Understand
Cannabis is one of the most commercially valuable crop plants on Earth, and until very recently, one of the least genetically characterized. While corn breeders have had access to dense genetic maps, validated QTLs for hundreds of traits, and genomic prediction models for decades, cannabis breeders have operated largely in the dark, making crosses based on phenotype, selecting based on visual assessment, and advancing genetics without understanding the molecular architecture of the traits they were trying to improve.
This is changing rapidly. The convergence of affordable next-generation sequencing, high-quality reference genomes, and the lifting of research restrictions has enabled a wave of genome-wide association studies in cannabis that are, for the first time, mapping the specific genetic loci that control the traits commercial growers care about most.
The implications for breeding programs are transformative. Every validated marker-trait association is a tool that converts breeding from probability to precision, from “this cross might produce high-THC offspring” to “this seedling carries the alleles associated with high THC production at three independent QTLs.”
How GWAS Works: From Whole Genomes to Specific Genes
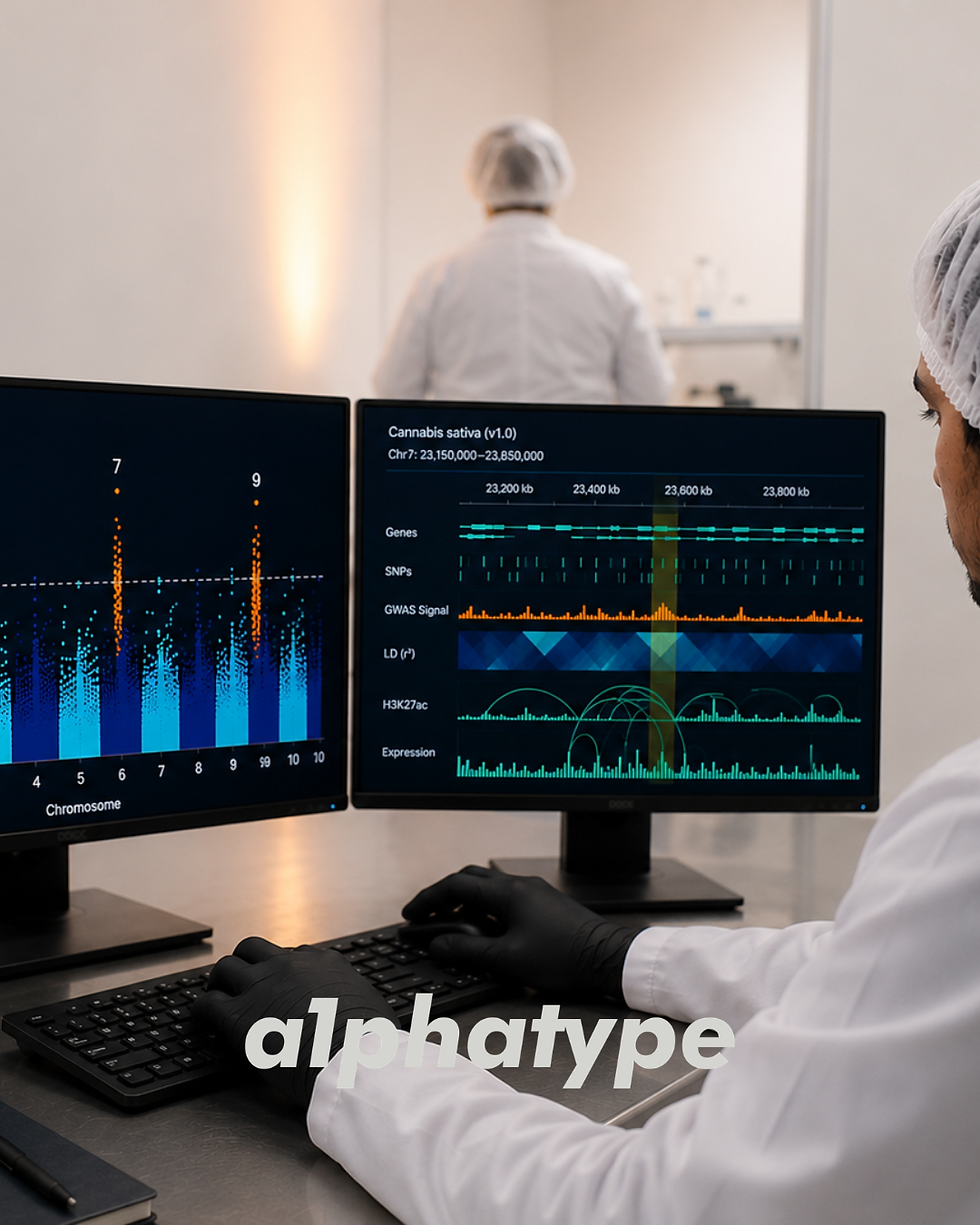
A genome-wide association study works by correlating genetic variation across the entire genome with variation in a phenotypic trait across a population of genetically diverse individuals. The logic is straightforward: if a specific DNA variant (a SNP) at a specific genomic position is found more frequently in high-THC plants than in low-THC plants, that variant is statistically associated with THC production, and the genomic region surrounding it likely contains a gene or regulatory element influencing the trait.
GWAS COMPONENT | WHAT IT INVOLVES | WHY IT MATTERS |
Diverse Association Panel | A population of 100–500+ genetically diverse cannabis accessions, representing the range of trait variation to be studied. Each individual is phenotyped for target traits and genotyped at hundreds of thousands of SNP markers. | Genetic diversity is the raw material of GWAS. The more diverse the panel, the more trait variation is available to map, and the finer the mapping resolution (because linkage disequilibrium decays faster in diverse populations). |
High-Density Genotyping | Each individual in the panel is genotyped using technologies like genotyping-by-sequencing (GBS), high-density GBS (HD-GBS), or SNP arrays, producing 100,000–300,000+ SNP markers distributed across all 10 cannabis chromosomes. | Dense marker coverage ensures that every genomic region is interrogated. The 2025 Canadian GWAS used 282,000 common SNPs—sufficient resolution to detect associations at sub-centimorgan scale. |
Phenotypic Measurement | Precise, quantitative measurement of target traits: HPLC/GC-MS for cannabinoids and terpenes, field measurements for yield and morphology, controlled inoculation assays for disease resistance. | GWAS is only as good as its phenotype data. Measurement error in the phenotype directly reduces statistical power to detect genetic associations. Replicated, multi-environment phenotyping is ideal. |
Statistical Association Testing | Mixed linear models (MLM) that account for population structure and kinship are used to test each SNP for association with each trait. Significance thresholds are adjusted for multiple testing (Bonferroni or FDR correction). | Controls for false positives caused by relatedness or population stratification—critical in cannabis, where commercial populations often share recent ancestry through common elite lineages. |
QTL Identification & Candidate Gene Analysis | Statistically significant markers define genomic regions (QTLs) associated with the trait. Genes within those regions are examined as candidates for the causal variant. Functional annotation connects markers to biological mechanisms. | Bridges the gap between statistical association and biological understanding. Identifying the causal gene enables development of diagnostic markers for breeding programs. |
What Cannabis GWAS Has Revealed So Far
The first generation of cannabis GWAS results is already reshaping how we understand the plant’s genetic architecture:
TRAIT CATEGORY | KEY GWAS FINDINGS | BREEDING IMPLICATION |
Cannabinoid Profiles | 33 significant markers associated with 11 cannabinoid traits in drug-type cannabis. A ~60 Mb haplotype on chromosome 7 is strongly associated with THC-dominant chemotype. Multiple QTLs on chromosomes 1, 4, 6, and 9 influence individual cannabinoid concentrations. | Chemotype can be predicted with high confidence from molecular markers. Total cannabinoid content is polygenic and requires multi-locus prediction models. |
Terpene Profiles | GWAS on drug-type cannabis identified QTLs associated with specific terpene compounds, confirming that individual terpenes have partially independent genetic control despite sharing biosynthetic precursor pathways. | Terpene profiles can be engineered through targeted marker-assisted selection for specific terpene QTLs, enabling breeders to design flavor profiles rather than just selecting for them. |
Flowering Time | Multiple QTLs identified across chromosomes, including loci near genes homologous to known flowering-time regulators in Arabidopsis (FT, CO, FLC pathway orthologs). Significant G×E interaction observed across environments. | Flowering time is genetically tunable. Markers enable selection for specific maturation windows matched to target environments (latitude, facility type). |
Sex Determination | QTLs on the sex chromosomes confirmed, with candidate genes involved in hormone balance (auxin and gibberellic acid pathways) identified in sex-determination regions. | Molecular sex markers validated through GWAS provide the genetic basis for early sex identification and intersex stability screening. |
Morphological Traits | Plant height, internode distance, branching pattern, stem diameter, and days to maturity all show significant marker-trait associations, confirming quantitative genetic control with multiple loci of moderate effect. | Architecture traits amenable to marker-assisted selection, enabling breeders to match plant morphology to specific facility types (indoor compact vs. outdoor vigorous). |
From GWAS to Genomic Selection: The Next Frontier
GWAS identifies individual loci with statistically significant effects on a trait. This works well for traits controlled by a few genes of large effect, chemotype ratio, sex determination, and autoflowering. But many commercially critical traits, total yield, overall cannabinoid concentration, broad-spectrum disease resistance, are polygenic: controlled by many genes, each contributing a small effect.
For polygenic traits, no single marker explains enough of the trait variation to be useful as a standalone selection tool. This is where genomic selection (GS) enters the picture.
Genomic selection uses all markers across the genome simultaneously to calculate a genomic estimated breeding value (GEBV) for each individual. This single number predicts how well that individual’s offspring will perform for the trait of interest. Instead of asking “does this plant carry the right allele at locus X?” (which is what MAS does), GS asks “what is this plant’s overall genetic merit across all loci that influence this trait?”
APPROACH | BEST FOR | LIMITATION |
Marker-Assisted Selection (MAS) | Traits controlled by one or a few genes of large effect: chemotype, sex, autoflowering, specific disease resistance alleles. Uses 1–10 markers per trait. | Cannot capture polygenic variation. For complex traits like yield, MAS based on a few markers leaves most of the genetic variation unexplained. |
Genomic Selection (GS) | Polygenic traits controlled by many genes of small effect: total yield, overall cannabinoid concentration, broad stress tolerance, general combining ability. Uses thousands of markers simultaneously. | Requires a training population with both genotype and phenotype data to build the prediction model. Model accuracy depends on training population size, marker density, and genetic relatedness between training and selection populations. |
Combined MAS + GS | Programs targeting both simple and complex traits simultaneously: fix chemotype with MAS while improving yield with GS in the same breeding cycle. | Highest data and computational requirements. But provides the most comprehensive selection capability available. |
Genomic selection has transformed breeding in corn, wheat, dairy cattle, and forestry—increasing genetic gain per year by 2–3x compared to conventional phenotypic selection. Cannabis is now at the threshold of adopting this technology, and the programs that build the training populations and prediction models first will have a structural advantage that compounds with every breeding cycle.
Building the Infrastructure: What GS Requires
Implementing genomic selection in cannabis requires three foundational investments:
GS Infrastructure Requirements
A genotyped and phenotyped training population. Hundreds of individuals representing the genetic diversity of the breeding program, each genotyped at high marker density and phenotyped for target traits under replicated, multi-environment conditions. This training population is the dataset from which the GS prediction model is built.
Statistical modeling capacity. Genomic BLUP (GBLUP), Bayesian regression models, or machine learning approaches that can estimate the effect of every marker across the genome simultaneously. These models are computationally intensive but well-established in other crop species.
A breeding pipeline that can act on GEBVs. Once GEBVs are calculated for a population of selection candidates, the breeding program must be able to select and cross the top-ranked individuals rapidly, which is where speed breeding and molecular sex determination become essential complementary technologies.
What This Means for the Industry
The transition from phenotype-based selection to genomics-informed selection is the single largest methodological shift in the history of cannabis breeding. It is not optional for programs that intend to remain competitive over the next decade.
Every other commercial crop has made this transition. Corn breeders select on genomic breeding values. Dairy geneticists select bulls based on GEBVs before those bulls ever sire a calf. Forest tree breeders use genomic prediction to evaluate seedlings years before they produce wood. The programs that adopted these tools first now dominate their industries. The same dynamic will play out in cannabis.
For cultivators, this means that the genetics available from programs investing in GWAS and genomic selection will be measurably superior—not because of any single breakthrough, but because of the cumulative advantage of making better selection decisions, faster, at every generation of the breeding cycle.
Alphatype’s Genomics-First Breeding Model
Alphatype is building the genomic infrastructure that cannabis breeding has lacked. Our breeding populations are genotyped at high marker density. Our phenotyping programs generate replicated, multi-environment data for cannabinoid, terpene, yield, morphological, and disease resistance traits. And our breeding decisions integrate marker-assisted selection for major-effect loci with genomic prediction models for complex, polygenic traits.
We are not waiting for the rest of the industry to adopt genomic selection. We are building the training populations, validating the prediction models, and making selection decisions on breeding values today. The genetic gain this generates compounds with every cycle, and every cycle that passes without it is genetic gain permanently lost.





















































